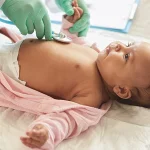
بیماری فنیل کتونوری یک اختلال متابولیکی ژنتیکی است که به دلیل نقص در آنزیم فنیل آلانین هیدروکسیلاز ایجاد میشود. این آنزیم مسئول تبدیل فنیل آلانین، یک آمینواسید ضروری، به تیروزین است. در افراد مبتلا به فنیل کتونوری، این آنزیم به درستی عمل نمیکند و در نتیجه فنیل آلانین در خون و بافتها تجمع مییابد. این تجمع به مغز آسیب زده و موجب مشکلات عصبی، عقبماندگی ذهنی و دیگر اختلالات جدی در رشد و تکامل فرد میشود. این بیماری بهطور معمول در دوران نوزادی تشخیص داده میشود، زیرا در بسیاری از کشورهای جهان، آزمایش غربالگری نوزادان برای فنیل کتونوری انجام میشود.
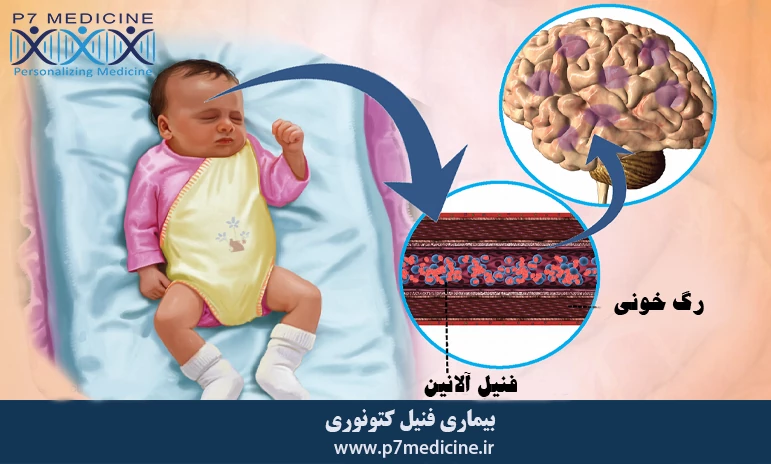
بیماری فنیل کتونوری چیست؟
بیماری فنیل کتونوری یک اختلال متابولیک ارثی است که در آن بدن قادر به تجزیه و پردازش فنیل آلانین، یک آمینواسید ضروری موجود در بسیاری از پروتئین ها نیست. این عدم توانایی به دلیل کمبود یا نقص در آنزیمی به نام فنیل آلانین هیدروکسیلاز اتفاق میافتد. در نتیجه، سطح فنیل آلانین در خون افزایش مییابد و به مغز آسیب میرساند. این بیماری در صورتی که به موقع تشخیص داده نشود و درمان نشود، منجر به مشکلات شدید ذهنی و جسمی مانند عقب ماندگی ذهنی، اختلالات رفتاری و مشکلات عصبی شود. درمان PKU شامل رعایت یک رژیم غذایی خاص با محدودیت شدید پروتئین های حاوی فنیل آلانین و استفاده از مکمل های غذایی است تا سطح این آمینو اسید در خون کنترل شود.
بیشتر بخوانید: ژن درمانی چیست؟
علائم بیماری فنیل کتونوری
علائم بیماری فنیل کتونوری معمولاً در دوران نوزادی یا کودکی ظاهر میشود و اگر درمان نشود، به مشکلات جدی در رشد و تکامل کودک منجر میشود. این علائم عبارتند از:
- تأخیر در رشد و توسعه ذهنی: یکی از علائم اصلی PKU، تأخیر در رشد ذهنی و اختلالات یادگیری است که اگر درمان نشود، منجر به عقب ماندگی ذهنی شدید میشود.
- مشکلات رفتاری و روانی: کودکانی که درمان نشوند، مشکلات رفتاری مانند پرخاشگری، بیقراری و اختلالات روانی از قبیل اضطراب و افسردگی را تجربه میکنند.
- اختلالات حرکتی و عصبی: افزایش فنیل آلانین در مغز باعث مشکلات حرکتی مانند حرکات غیرطبیعی یا کندی در حرکت میشود. در برخی موارد، این اختلالات به فلجی یا مشکلات عضلانی منجر میشود.
- بوی بد پوست و ادرار: نوزادان و کودکان مبتلا به PKU معمولاً بویی مشابه بوی ماده شیمیایی در پوست و ادرارشان دارند که ناشی از تجمع فنیل آلانین و محصولات متابولیک آن است.
- مشکلات در تغذیه: در بعضی موارد، کودکانی که مبتلا به بیماری فنیل کتونوری هستند، از تغذیه دچار مشکل میشوند و تمایل کمتری به خوردن غذاهای معمولی دارند.
- مشکلات گوارشی: برخی از کودکان مبتلا به این بیماری دچار استفراغ، اسهال، یا سایر مشکلات گوارشی میشوند.
بیشتر بخوانید: بیماری هانتینگتون
علل بیماری فنیل کتونوری
بیماری فنیل کتونوری یک اختلال ژنتیکی است که به علت نقص یا کمبود آنزیم فنیل آلانین هیدروکسیلاز ایجاد میشود. این آنزیم مسئول تبدیل فنیل آلانین، یک آمینو اسید موجود در پروتئین ها، به ترکیب دیگری به نام تیروزین است. زمانی که این آنزیم به درستی عمل نکند، فنیل آلانین در خون تجمع مییابد و میتواند به مغز آسیب برساند. علت اصلی بیماری فنیل کتونوری یک جهش ژنتیکی است که در هر دو والدین منتقل میشود. این بیماری به صورت اتوزوم مغلوب به ارث میرسد، به این معنی که برای بروز بیماری، هر دو والد باید ژن معیوب را به فرزند خود منتقل کنند. اگر تنها یکی از والدین ژن معیوب را داشته باشد، فرد حامل خواهد بود و علائم بیماری را تجربه نخواهد کرد، اما قادر به انتقال بیماری به نسل بعدی است. در نتیجه، علت اصلی فنیل کتونوری وجود جهش های ژنتیکی در هر دو کروموزوم ۱۲ است که باعث نقص در آنزیم فنیل آلانین هیدروکسیلاز میشود و مانع از پردازش صحیح فنیل آلانین در بدن میگردد.
تشخیص بیماری فنیل کتونوری
تشخیص بیماری فنیل کتونوری معمولاً از طریق آزمایش خون نوزادان در هفته اول زندگی انجام میشود. این آزمایش، که به نام آزمایش غربالگری نوزادی شناخته میشود، برای شناسایی سطح فنیل آلانین در خون نوزاد استفاده میشود. در صورتی که سطح فنیل آلانین بالاتر از حد طبیعی باشد، احتمال ابتلا به فنیل کتونوری بررسی میشود و آزمایش های تکمیلی مانند آزمایش های ژنتیکی یا آنزیمی برای تأیید تشخیص انجام میشود. این آزمایش ها وجود نقص در آنزیم فنیل آلانین هیدروکسیلاز را نشان میدهند و بیماری را به طور قطعی تأیید میکنند.
بیشتر بخوانید: آیا سرطان سینه ارثی است؟
درمان بیماری فنیل کتونوری
درمان بیماری فنیل کتونوری معمولاً شامل رعایت یک رژیم غذایی خاص و محدودیت مصرف فنیل آلانین است. این درمان به طور عمده بر کنترل سطح فنیل آلانین در خون متمرکز است تا از آسیب های مغزی و مشکلات ذهنی جلوگیری شود. در اینجا برخی از روش های درمانی بیماری PKU آورده شده است:
رژیم غذایی خاص:
اصلیترین روش درمان PKU، رعایت رژیم غذایی با محدودیت شدید فنیل آلانین است. بیماران باید از مصرف مواد غذایی حاوی پروتئین های حیوانی و گیاهی که غنی از فنیل آلانین هستند، مانند گوشت، تخم مرغ، شیر و برخی غلات، خودداری کنند. در عوض، مصرف مواد غذایی با پروتئین های کم فنیل آلانین مانند میوه ها، سبزیجات و برخی مواد غذایی ویژه برای بیماران فنیل کتونوری توصیه میشود.
استفاده از مکمل های فنیل آلانین:
برای جبران کمبود پروتئین های ضروری، بیماران باید از مکمل های پروتئینی مخصوصی که فاقد فنیل آلانین هستند، استفاده کنند. این مکمل ها به تأمین نیاز بدن به پروتئین بدون افزایش سطح فنیل آلانین در خون کمک میکنند.
داروهای کمکی:
برخی از داروها برای کمک به متابولیسم فنیل آلانین و کاهش سطح آن در خون تجویز میشوند. یکی از این داروها کودرنس است که به بهبود عملکرد آنزیم فنیل آلانین هیدروکسیلاز کمک میکند.
پایش منظم سطح فنیل آلانین:
برای اطمینان از کنترل مناسب بیماری، بیماران باید به طور منظم سطح فنیل آلانین در خون خود را تحت نظر پزشک قرار دهند و رژیم غذایی و درمان های خود را بر اساس نتایج آزمایش ها تنظیم کنند.
سوالات متداول
بیماری فنیل کتونوری درمان پذیر است؟
در حال حاضر، بیماری فنیل کتونوری درمان قطعی ندارد، اما با رعایت رژیم غذایی خاص و محدودیت مصرف فنیل آلانین، بیماران از بروز مشکلات شدید مغزی و ذهنی جلوگیری میکنند و زندگی سالمی خواهند داشت.
بیماری فنیل کتونوری ارثی است؟
بیماری فنیل کتونوری یک اختلال ژنتیکی است که به صورت اتوزوم مغلوب به ارث میرسد. به این معنی که هر دو والد باید ژن معیوب را منتقل کنند تا بیماری در فرزند ظاهر شود.
بیماران مبتلا به فنیل کتونوری میتوانند زندگی عادی داشته باشند؟
اگر بیماری به موقع تشخیص داده شود و درمان های مناسب از جمله رژیم غذایی خاص رعایت شود، افراد مبتلا به PKU زندگی عادی خواهند داشت.
نتیجه گیری
بیماری فنیل کتونوری یک اختلال ژنتیکی است که با تشخیص و درمان به موقع میتوان از بروز مشکلات جدی و دائمی مغزی و ذهنی پیشگیری کرد. رعایت رژیم غذایی خاص و محدودیت مصرف فنیل آلانین برای بیماران ضروری است و با نظارت دقیق بر سطح فنیل آلانین، این بیماران زندگی سالم و طبیعی خواهند داشت. اهمیت تشخیص زودهنگام و پیگیری های پزشکی مستمر در این بیماری بر هیچ کس پوشیده نیست و توجه به این نکات کیفیت زندگی افراد مبتلا به PKU را بهبود میبخشد.